SHAPEwarp uses an approach inspired by the BLAST heuristic algorithm to identify regions of RNA structure similarity in a database of chemical reactivity profiles generated by SHAPE (Selective 2'-hydroxyl acylation analyzed by primer extension).
SHAPEwarp Workflow
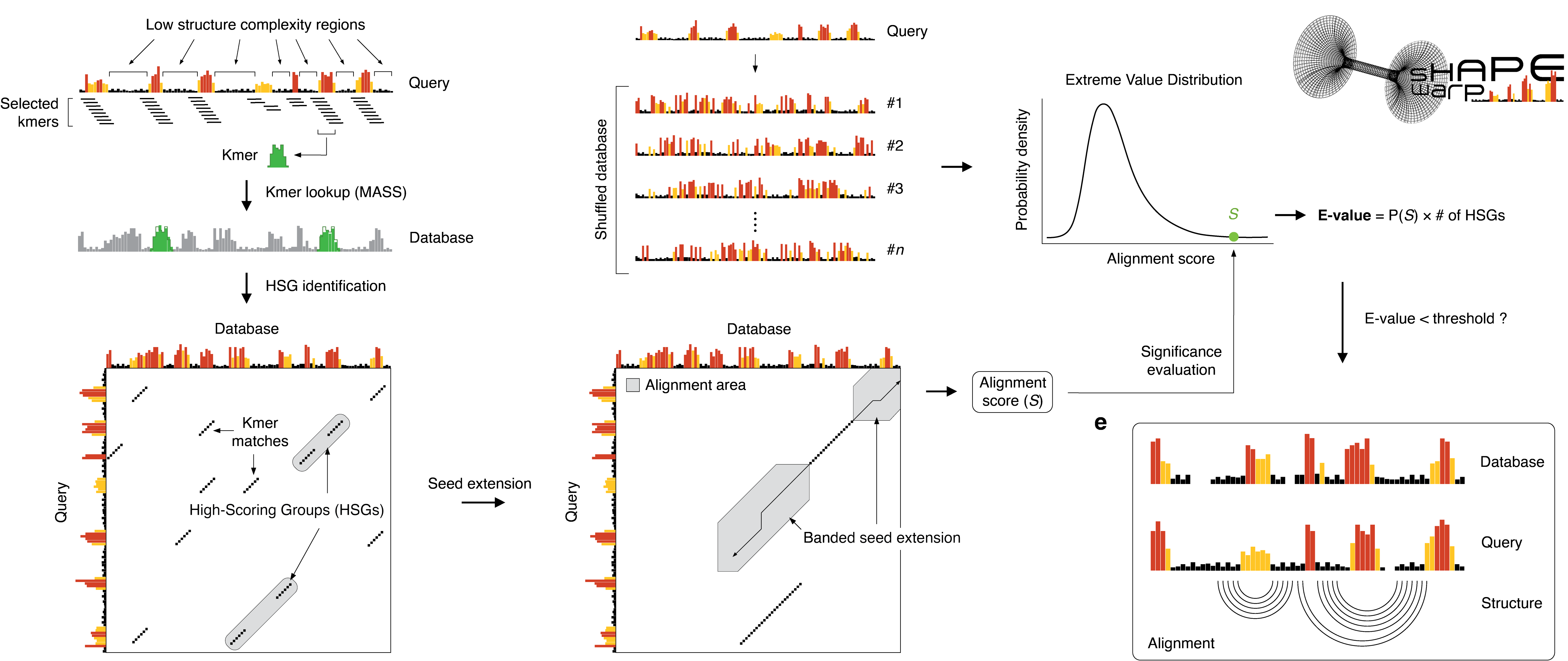
Given a user-provided query, the following analysis steps are performed:
{kind=link}
-
All the possible query kmers of a user-defined length are enumerated, filtering out those corresponding to low structure complexity regions (i.e. those characterized by a Gini coefficient below a certain cutoff).
-
Kmers passing the complexity cutoff are then looked up in a database of reactivity profiles via MASS, a highly optimized algorithm for time series subsequences all-pairs-similarity search (TSAPSS) in just O(n2). Additionally, the percent GC content of the kmer and its matches is evaluated and matches having GC content deviating by more than a certain percentage from that of the query kmer, are discarded.
-
Kmer matches lying on the same diagonal, within a maximum distance from each other, are then grouped into high scoring groups (HSGs).
-
Ungapped HSGs are extended upstream and downstream by using a banded semi-global alignment algorithm, that incorporates both features of the Gotoh’s Smith-Waterman algorithm with affine gap penalties and of DTW. The extension process is stopped when the score drops below a certain threshold for more than a certain number of residues.
-
A null model is then built by searching the same query in a database of randomly shuffled reactivity profiles and used to estimate the E-value of each database match.
-
For those matches passing the user-defined E-value threshold, the aligned reactivity profiles can then be used to model the underlying structure by using the RNAalifold algorithm of the ViennaRNA package.
Usage
To list the required parameters, simply type:
$ SHAPEwarp -h
| Parameter | Type | Description |
|---|---|---|
| -db or --database | string | Path to a database file, or to a (directory of) RNA Framework XML file(s) |
| --shuffled-db | string | Path to a shuffled database file Note: if not provided, shuffling will be performed on the fly |
| --dump-db | string | Dumps the imported XML files to the specified database file |
| --dump-shuffled-db | string | Dumps the shuffled database to the specified file |
| --db-shuffles | int | Number of shuffles to perform for each sequence in the database (Default: 100) Note: see Building the database section below) |
| --db-block-size | int | Size (in nt) of the blocks for shuffling the sequences in the database (Default: 10) |
| --db-in-block-shuffle | Besides shuffling blocks, residues within each block will be shuffled as well | |
| -q or --query | string | Path to the query file Note: each entry should contain (one per row) the sequence id, the nucleotide sequence and a comma-separated list of SHAPE reactivities (see SHAPEwarp query files section below) |
| -o or --output | string | Output directory (Default: sw_out/) |
| --overwrite | Overwrites the output directory (if the specified path already exists) | |
| --threads | int | Number of processors to use (Default: 1) |
| --max-reactivity | float | Maximum value to which reactivities will be capped (Default: 1) |
| --max-align-overlap | float | If two significant alignments overlap by more than this value, the least significant one (the one with the lowest alignment score) will be discarded (Default: 0.5) |
| --null-hsgs | int | Number of HSGs in the shuffled database to be extended to build the null model (Default: 10000) |
| --inc-e or --inclusion-evalue | float | E-value threshold to consider an alignment significant (Default: 0.01) |
| --rep-e or --report-evalue | float | E-value threshold to report a match (Default: 0.1 |
| --report-alignment | string | Reports sequence alignments in the specified format (fasta or stockholm) Note: alignments are reported only for matches below the inclusion E-value cutoff |
| --report-reactivity | Reports the aligned reactivities for significant matches in the reactivities/ subfolder of the output directory, in JSON format |
|
| Kmer lookup options | ||
| --min-kmers | int | Minimum number of kmers required to form a High Scoring Group (HSG; Default: 2) |
| --max-kmer-dist | int | Maximum distance between two kmers to be merged in a HSG (Default: 30) |
| --kmer-len | int | Length (in nt) of the kmers (Default: 15) |
| --kmer-offset | int | Sliding offset for extracting candidate kmers from the query (Default: 1) |
| --match-kmer-gc-content | The sequence of a query kmer and the corresponding database match must have GC% contents differing no more than --kmer-max-gc-diff |
|
| --kmer-max-gc-diff | float | Maximum allowed GC% difference to retain a kmer match (requires --match-kmer-gc-content)Note: the default value is automatically determined based on the chosen kmer length |
| --match-kmer-seq | The sequence of a query kmer and the corresponding database match must differ no more than --kmer-max-seq-dist |
|
| --kmer-max-seq-dist | float | Maximum allowed sequence distance to retain a kmer match (requires --match-kmer-seq; Default: 0)Note: when >= 1, this is interpreted as the absolute number of bases that are allowed to differ between the kmer and the matching region. When < 1, this is interpreted as a fraction of the kmer's length |
| --kmer-min-complexity | float | Minimum complexity (measured as Gini coefficient) of candidate kmers (Default: 0.3) |
| --kmer-max-match-every-nt | int | A kmer is allowed to match a database entry on average every this many nt (Default: 200) Note: this value is automatically adjusted for shorter database entries |
| Alignment options | ||
| --align-match-score | float,float | Minimum and maximum score reactivity differences below 0.5 will be mapped to (Default: 0,2) |
| --align-mismatch-score | float,float | Minimum and maximum score reactivity differences above 0.5 will be mapped to (Default: -6,-0.5) |
| --align-gap-open-penalty | float | Gap open penalty (Default: -14) |
| --align-gap-ext-penalty | float | Gap extension penalty (Default: -5) |
| --align-max-drop-off-rate | float | An alignment is allowed to drop by maximum this fraction of the best score encountered so far, before extension is interrupted (Default: 0.8) |
| --align-max-drop-off-bases | int | An alignment is allowed to drop below the best score encountered so far * --align-max-drop-off-rate by this number of bases, before extension is interrupted (Default: 8) |
| --align-len-tolerance | float | The maximum allowed tollerated length difference between the query and db sequences to look for the ideal alignment along the diagonal (measured as a fraction of the length of the shortest sequence between the db and the query) (Default: 0.1) |
| --align-score-seq | Sequence matches are rewarded during the alignment | |
| --align-seq-match-score | float | Score reward for matching bases (Default: 0.5) |
| --align-seq-mismatch-score | float | Score reward for matching bases (Default: -2) |
| Alignment folding evaluation options | ||
| --eval-align-fold | Alignments passing the --inclusion-evalue threshold, are further evaluated for the presence of a conserved RNA structure by using RNAalifold |
|
| --shuffles | Number of shuffles to perform for each alignment (Default: 100) | |
| --block-size | int | Size (in nt) of the blocks for shuffling the alignment (Default: 3) |
| --in-block-shuffle | Besides shuffling blocks, residues within each block will be shuffled as well | |
| --min-bp-support | float | Minimum fraction of base-pairs of the RNAalifold-inferred structure that should be supported by both query and db sequence to retain a match (Default: 0.75) |
| --ribosum-scoring | Use RIBOSUM scoring matrix | |
| --slope | float | Slope for SHAPE reactivities conversion into pseudo-free energy contributions (Default: 1.8) |
| --intercept | float | Intercept for SHAPE reactivities conversion into pseudo-free energy contributions (Default: 1.8) |
| --max-bp-span | int | Maximum allowed base-pairing distance (Default: 600) |
| --no-lonely-pairs | Disallows lonely pairs (helices of 1 bp) | |
| --no-closing-gu | Disallows G:U wobbles at the end of helices | |
| --temperature | float | Folding temperature in Celsius (Default: 37.0) |
Building the database
Since version 2.0.0, in addition to standard binary database files, SHAPEWarp can directly import (directories of) SHAPE XML reactivity files (generated using RNA Framework; check the documentation for additional details). Similarly, if no shuffled database is provided, database shuffling will be performed on the fly.
However, when working with large datasets, it might be convenient to build and store a binary database file. This can be accomplished as it follows:
$ SHAPEwarp --database /path/to/directory/of/XML/files --dump-db /path/to/database.db
Similarly, to reduce runtimes, or to ensure reproducible search results, it might be convenient to dump the shuffled database to file:
$ SHAPEwarp --database /path/to/directory/of/XML/files --dump-db /path/to/binary/database.db --dump-shuffled-db /path/to/shuffled/database.db
By default, 100× shuffles (--db-shuffles) of the original database will be performed. In most cases such a high number of shuffles is not needed and it will only exponentially increase the search time. As a rule of thumb, the number of shuffles needed can be determined using the following empirical formula:
where L is the sum of the lengths of all the sequences in the database.
Understanding the algorithm
While it is advisable for most users to run SHAPEwarp with its default parameters, as these are the results of a careful and thorough calibration, it might be useful to adjust the analysis on a case-by-case basis.
First of all, as SHAPE reactivities can have a variable range, depending on the normalization scheme that has been adopted, both query and database reactivities are capped to a maximum value of max-reactivity. During the kmer lookup phase, the algorithm enumerates all the possible kmers of size kmer-len in the user-provided query, filtering out those:
- encompassing NaN values
- having Gini coefficient <
kmer-min-complextiy, corresponding to regions of low structural complexity (expected to match most transcripts)
Kmers are matched against the database using the MASS algorithm. Given a kmer and a database reactivity profile, MASS returns an array of distances of that kmer to each position of the database profile. Matches having a distance < μ - 3 × σ, where μ and σ are respectively the mean and the standard deviation of the distances, are retained. Kmer-match pairs are then filtered out on the basis of the following criteria:
- when
match-gc-contentis enabled and the difference between the GC% content of the kmer and that of its database match is >kmer-max-gc-diff - when
match-kmer-seqis enabled and the sequence of the kmer and that of its database match differ by more thanmax-kmer-seq-dist - when the ratio between the length of the database transcript and the number of matches for a given kmer in that transcript is <
kmer-max-match-every-nt
The retained kmers are then grouped into high-scoring groups (HSGs). HSGs are made of kmers residing on the same diagonal (so, corresponding to regions of the query and database that can be theoretically aligned without opening gaps). Each HSG is made of ≥ min-kmers kmers, residing within a maximum distance of max-kmer-dist from each other.
Each HSG constitutes the seed from which an alignment extension will be attempted, both upstream and downstream. The seed extension uses a dynamic programming algorithm that incorporates features of both dynamic time warping (DTW) and of Gotoh's Smith-Waterman implementation with affine gap penalties. In brief, if the reactivity difference between two bases is < 0.5, the corresponding alignment score is calculated by linearly mapping the reactivity difference to the align-match-score range. Similarly, if the reactivity difference is ≥ 0.5, the corresponding alignment score is calculated by linearly mapping the reactivity difference to the align-mismatch-score range. Affine gap penalties are controlled via the align-gap-open-penalty, the gap open penalty, and align-gap-ext-penalty, the gap extension penalty. During the matrix-filling stage of the algorithm, the alignment score is allowed to drop by not more than align-max-drop-off-rate × the best alignment score encountered so far, for no more than align-max-drop-off-bases. While the algorithm is sequence-agnostic by default, sequence can be optionally taken into account by enabling the align-score-seq option. When enabled, sequence matches are rewarded by align-seq-match-score and mismatches are penalized by align-seq-mismatch-score. To further speed-up the alignment phase, the alignment matrix is only filled in a band of size b around the diagonal, where b is defined as min(10, align-len-tolerance × min(q, d)), where q and d are respectively the length of the query and of the database entry. Therefore, a minimum band size of 10 nt is always granted. The final alignment score S is calculated as the sum of the scores of the HSG (the seed), plus the two upstream and downstream extensions.
After having extended any possible HSG in the database, the same procedure is repeated by searching the query in a database of shuffled reactivity profiles. A maximum of null-hsgs HSGs identified in the shuffled database are extended, and the resulting alignment scores are used to build the null model, hence allowing to calculate a p-value for each alignment from the real database. The p-value is the probability of identifying by chance a match having a score ≥ S. The E-value corresponds to the number of expected database matches having a score ≥ S and it is thus calculated as p-value × the number of extended HSGs. It might happen, especially with long queries, that multiple HSGs will be identified for the same query-database entry pair, hence resulting in multiple overlapping alignments. To prevent duplicate matches from being reported, if multiple alignments overlap by more than max-align-overlap, only the one with the lowest E-value is reported.
Optionally, if the eval-align-fold option is enabled, alignments passing the inc-e E-value cutoff can be evaluated for the presence of a consensus secondary structure, via RNAalifold. RNAalifold uses both the sequence alignment and the corresponding SHAPE reactivity information. SHAPE reactivities are converted into pseudo free energy contributions by using the approach from Deigan et al., 2009:
ΔGSHAPE(i) = slope · ln[Reactivity(i) + 1] + intercept
If less than min-bp-support base-pairs from the predicted consensus structure are supported for either of the two aligned RNAs, the match is discarded. Furthermore, the probability of predicting a structure with a free energy ≤ to that of the consensus structure inferred from the alignment is evaluated by randomly shuffling shuffles times the columns of the original alignment in block-size-long blocks, followed by RNAalifold analysis. A z-score is calculated and the corresponding p-value determined. If the p-value is < 0.05, the alignment and the inferred structure are reported.
SHAPEwarp query files
SHAPEwarp query files are plain-text files, containing the unique identifier, nucleotide sequence and reactivity profile, one per line, for each query to be searched against the database. The typical SHAPEwarp query file has the following structure:
Query_1
AAATTGAAGAGTTTGATCATGGCTCAGATTGAACGCTGGCGGCAGGCCTAACACATGCAAGTCGAACGGTAACAGGAAGAAGCTTGCTTCTTTGCTGACGAGTGGCGGACGGGTGAGTAATGTCTGGGAAACTGCCTGATGGAGGGGGATAACTACTGGAAACGGTAGCTAATACCGCATAACGTCGCAAGACCAAAGAG
NaN,NaN,NaN,NaN,NaN,NaN,NaN,NaN,NaN,NaN,0.172,0.017,0.034,0.224,0.301,0.135,0.513,0.866,0.816,1.305,0.267,0.149,0.023,0.160,0.226,0.130,0.389,0.114,0.082,0.299,0.803,0.034,0.047,0.046,0.426,0.309,0.603,1.189,0.216,0.067,0.048,0.047,0.055,0.056,0.027,0.348,0.098,0.313,0.887,0.631,0.119,0.322,0.045,0.379,0.604,0.889,0.341,0.286,0.247,0.780,0.816,0.698,0.494,0.877,0.481,0.483,NaN,0.814,0.624,0.880,0.286,0.382,0.141,0.148,0.125,0.226,0.051,0.075,NaN,NaN,0.070,0.278,0.826,1.836,2.690,0.802,0.648,0.619,NaN,NaN,0.273,0.270,NaN,1.163,0.081,0.163,0.466,0.259,0.283,0.539,0.490,0.327,0.610,1.171,2.621,0.453,0.142,0.165,0.156,0.308,0.361,0.537,0.571,0.545,0.492,0.704,0.514,1.624,1.210,2.453,3.827,0.433,0.216,0.024,0.046,0.013,0.028,0.116,0.176,0.638,NaN,0.281,0.808,0.595,0.085,0.103,0.278,0.083,0.056,0.165,0.049,0.267,0.471,0.282,0.772,0.068,0.069,0.295,0.058,0.506,0.359,0.157,0.090,0.180,0.140,0.098,0.166,0.124,0.186,0.173,0.352,0.392,0.197,0.079,0.072,0.013,0.027,0.040,0.081,0.151,0.272,0.451,1.250,0.953,0.045,0.151,2.557,0.899,0.667,1.237,0.914,0.609,NaN,0.042,0.074,0.108,0.459,1.154,0.419,0.498,0.113,0.053,0.100,0.364,0.157,0.487,0.608,2.693,0.679,0.306
Query_2
AGTGGCGGACGGGTGAGTAATGTCTGGGAAACTGCCTGATGGAGGGGGATAACTACTGGAAACGGTAGCTAATACCGCATAACGTCGCAAGACCAAAGAGGGGGACCTTCGGGCCTCTTGCCATCGGATGTGCCCAGATGGGATTAGCTAGTAGGTGGGGTAACGGCTCACCTAGGCGACGATCCCTAGCTGGTCTGAGA
0.490,0.327,0.610,1.171,2.621,0.453,0.142,0.165,0.156,0.308,0.361,0.537,0.571,0.545,0.492,0.704,0.514,1.624,1.210,2.453,3.827,0.433,0.216,0.024,0.046,0.013,0.028,0.116,0.176,0.638,NaN,0.281,0.808,0.595,0.085,0.103,0.278,0.083,0.056,0.165,0.049,0.267,0.471,0.282,0.772,0.068,0.069,0.295,0.058,0.506,0.359,0.157,0.090,0.180,0.140,0.098,0.166,0.124,0.186,0.173,0.352,0.392,0.197,0.079,0.072,0.013,0.027,0.040,0.081,0.151,0.272,0.451,1.250,0.953,0.045,0.151,2.557,0.899,0.667,1.237,0.914,0.609,NaN,0.042,0.074,0.108,0.459,1.154,0.419,0.498,0.113,0.053,0.100,0.364,0.157,0.487,0.608,2.693,0.679,0.306,0.124,0.103,0.209,0.631,1.584,0.590,0.836,NaN,1.426,2.272,0.990,0.501,0.437,0.072,0.056,0.117,0.116,0.165,0.582,0.407,0.007,0.029,0.034,0.031,0.048,NaN,0.127,0.236,0.447,0.212,0.331,0.386,0.041,0.051,0.004,0.058,0.073,0.165,0.735,0.025,0.055,0.145,0.604,0.394,0.898,0.306,0.121,0.170,0.557,NaN,1.113,1.024,NaN,0.779,0.025,0.086,0.029,0.163,0.020,0.121,0.228,0.389,0.666,0.485,0.900,0.385,0.044,0.033,0.011,0.026,0.052,0.000,NaN,0.309,0.023,0.066,0.088,0.076,0.641,0.954,0.327,0.174,0.108,0.046,0.012,0.069,0.189,0.310,0.073,0.030,0.064,0.058,0.182,0.110,0.197,0.362,1.155,1.123,0.400,1.363
SHAPEwarp output
The typical SHAPEwarp's output has the following structure:
Query DB entry Qstart Qend Dstart Dend Qseed Dseed Score P-value E-value
16S_600 16S 0 199 608 807 60-114 668-722 190.53 4.47e-11 6.39e-09 !
16S_500 16S 0 196 508 704 160-186 668-694 203.47 2.30e-10 3.06e-08 !
16S_200 16S 9 199 216 406 120-183 327-390 187.13 3.13e-10 3.41e-08 !
16S_700 16S 0 161 708 869 65-129 773-837 159.63 3.19e-09 5.07e-07 !
16S_1300 16S 0 180 1308 1488 10-112 1318-1420 199.02 3.94e-09 6.19e-07 !
16S_900 16S 7 170 916 1079 16-100 925-1009 173.76 2.23e-08 1.16e-06 !
16S_400 16S 19 199 427 607 97-144 505-552 159.77 1.09e-07 1.42e-05 !
16S_300 16S 4 194 311 501 20-83 327-390 146.14 2.70e-07 3.40e-05 !
16S_800 16S 0 199 808 1008 73-199 882-1008 137.66 5.85e-07 6.67e-05 !
16S_1100 16S 49 199 1157 1307 68-96 1176-1204 132.35 9.51e-07 9.70e-05 !
16S_0 16S 10 161 17 168 85-102 92-109 87.73 6.57e-05 4.66e-03 !
16S_1200 16S 0 199 1208 1407 110-199 1318-1407 131.31 3.52e-05 5.39e-03 !
16S_1100 16S 40 194 55 209 170-185 185-200 80.03 4.52e-04 0.05 ?
16S_400 23S 25 155 1252 1382 74-92 1301-1319 87.09 3.55e-04 0.05 ?
16S_800 23S 47 164 1608 1725 64-81 1625-1642 80.20 4.64e-04 0.05 ?
16S_800 16S 46 161 30 145 56-71 40-55 76.82 6.86e-04 0.08 ?
where:
| Field | Description |
|---|---|
| Query | Query ID |
| DB entry | Database entry's ID |
| Qstart | First aligned base of the query |
| Qend | Last aligned base of the query |
| Dstart | First aligned base of the database entry |
| Dend | Last aligned base of the database entry |
| Qseed | HSG position in the query |
| Dseed | HSG position in the database entry |
| Score | Alignment score |
| P-value | The probability of finding by chance an alignment with the same (or greater) score |
| E-value | The number of expected matches by chance, with the same (or greater) score, in a database of the same size |
If the --eval-align-fold option has been enabled, three additional fields will be reported:
| Field | Description |
|---|---|
| TargetBpSupport | Fraction of base-pairs in the consensus structure, that are supported by the database entry's sequence |
| QueryBpSupport | Fraction of base-pairs in the consensus structure, that are supported by the query sequence |
| MfePvalue | The probability of obtaining by chance a consensus structure with the same (or lower) free energy |